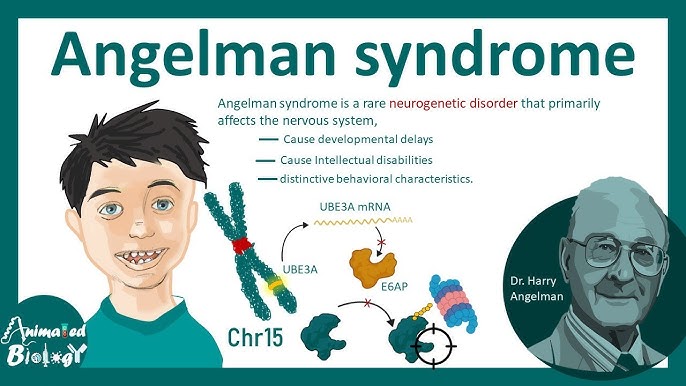
Angelman syndrome is a rare genetic disorder caused by changes in the UBE3A gene on chromosome 15. This condition affects the nervous system and is characterized by developmental delays, problems with speech and movement, intellectual disabilities, and in some cases, seizures. Despite these challenges, individuals with Angelman syndrome often exhibit a cheerful demeanor, with frequent smiling and laughter.
While there is no cure, early intervention and targeted treatments can help manage the symptoms and improve the quality of life for those affected.
Key Symptoms of Angelman Syndrome
The symptoms of Angelman syndrome typically become apparent between 6 to 12 months of age. These include:
Developmental and Neurological Symptoms:
- Developmental delays: Lack of crawling or babbling during infancy.
- Intellectual disability: Significant cognitive impairments.
- Speech challenges: Limited or absent verbal communication.
- Movement and balance issues: Difficulty walking and maintaining balance, often with stiff or jerky movements.
- Seizures: Begin around ages 2 to 3, often requiring medical management.
Behavioral and Physical Characteristics:
- Frequent smiling and laughter, often unrelated to external stimuli.
- Hyperexcitability: Easy to excite with increased hand-flapping and arm movements.
- Sleep disturbances: Difficulty falling asleep and staying asleep.
- Light-colored skin, hair, and eyes, in some cases due to reduced pigmentation.
Additional Symptoms:
- Feeding difficulties in infancy due to poor sucking and swallowing.
- Strabismus (crossed eyes).
- Scoliosis (curved spine) in older children or adults.
- Obesity, more common in older children and adults.
When to See a Doctor
Parents may first notice developmental delays, such as the absence of crawling or babbling, around 6 to 12 months of age. If these delays or other symptoms, such as seizures or movement difficulties, are present, consult a healthcare professional immediately for evaluation.
Causes of Angelman Syndrome
Angelman syndrome results from genetic changes affecting the maternal copy of the UBE3A gene, which is critical for brain development. Causes include:
- Missing or damaged maternal gene: The most common cause.
- Paternal uniparental disomy (UPD): Both chromosome 15 copies are inherited from the father, leaving the maternal gene inactive.
- Mutations in the UBE3A gene: Prevent the gene from functioning properly.
In most cases, these genetic changes occur spontaneously and are not inherited. However, in rare instances, the condition can be passed from an affected parent to a child.
Risk Factors
- Sporadic cases: Most individuals with Angelman syndrome have no family history of the condition.
- Inherited cases: Rarely, Angelman syndrome can be passed down through genetic mutations, increasing the risk in families with a history of the disorder.
Complications Associated with Angelman Syndrome
Individuals with Angelman syndrome may experience the following complications:
- Feeding difficulties:
- Poor coordination between sucking and swallowing in infants.
- May require high-calorie formulas for proper weight gain.
- Hyperactivity:
- Short attention spans and constant movement.
- Decreases with age and often does not require medication.
- Sleep disturbances:
- Irregular sleep-wake cycles and reduced sleep needs.
- Behavioral therapies and medications may improve sleep patterns.
- Orthopedic issues:
- Scoliosis may develop over time, necessitating monitoring and treatment.
- Obesity:
- Common in older children and adults due to reduced mobility and overeating tendencies.
- Seizures:
- Often require anti-seizure medications for management.
Diagnosis
Diagnosis typically involves a combination of:
- Developmental assessment: Identifying delays in milestones.
- Genetic testing: Detects abnormalities in the UBE3A gene.
- Clinical observation: Recognizes hallmark features, such as frequent laughter and movement difficulties.
Treatment and Management
While Angelman syndrome has no cure, treatments focus on managing symptoms and improving the individual’s overall quality of life. These include:
Medical Interventions:
- Seizure management: Anti-epileptic medications to control seizures.
- Sleep aids: Behavioral strategies and medications to address sleep disturbances.
Therapies:
- Physical therapy: Improves movement, balance, and coordination.
- Occupational therapy: Develops skills for daily living.
- Speech therapy: Focuses on nonverbal communication methods, such as sign language or augmentative communication devices.
Educational Support:
- Early intervention programs to address developmental delays.
- Tailored educational plans to support learning and social development.
Lifestyle Adjustments:
- High-calorie diets for infants with feeding difficulties.
- Monitoring for obesity and promoting physical activity.
Prevention and Genetic Counseling
- Since most cases are not inherited, prevention is generally not possible.
- For families with a history of Angelman syndrome, genetic counseling can provide valuable insights into the risk of recurrence in future pregnancies.
Outlook for Individuals with Angelman Syndrome
While Angelman syndrome affects cognitive and physical abilities, individuals often lead happy lives with appropriate support. Most people with Angelman syndrome live close to a typical lifespan. With advancements in therapy and medical care, they can achieve significant developmental milestones and enjoy meaningful relationships.
Conclusion
Angelman syndrome is a rare genetic condition with unique challenges and distinctive characteristics. Early diagnosis and a multidisciplinary approach to treatment can greatly enhance the quality of life for those affected. Families should seek regular support from healthcare providers, therapists, and support groups to navigate the complexities of the condition effectively.






{kind=link}